Qualität und Zulassung
Wer ist für die Zulassung von Biosimilars in der EU zuständig?
Biosimilars werden von der Europäischen Arzneimittelagentur (EMA) nach strengen Kriterien auf Qualität, Wirksamkeit und Sicherheit geprüft.
Damit ein Biosimilar zugelassen wird, muss nachgewiesen werden, dass es mit dem Referenzprodukt vergleichbar ist, und zwar durch:
- Vergleich der Arzneimittel-Qualität
- Vergleich der präklinischen Eigenschaften
- Vergleich der klinischen Eigenschaften
Gibt es Unterschiede zwischen Original (Referenzarzneimittel) und Biosimilar?
Ein Biosimilar muss in Aufbau und Wirkung dem Referenzarzneimittel so ähnlich sein, dass im Labor keine relevanten Unterschiede festgestellt werden können. Die Gleichwertigkeit zum Referenzarzneimittel in Bezug auf Qualität, biologische Aktivität, Sicherheit und Wirksamkeit muss durch einen umfangreichen direkten Vergleich (sogenanntes „Comparability Exercise“) nachgewiesen werden. Im Vergleich zum Referenzarzneimittel darf sich die Wirkung und Struktur eines Biosimilars nur innerhalb bestimmter Grenzen, der sogenannten Schwankungsbreite bewegen. Eine gewisse Schwankungsbreite ist jedoch unvermeidbar, da es zwischen zwei Chargen desselben biologischen Arzneimittels keine exakte Übereinstimmung geben kann. Dies trifft auch auf das Original-Biopharmazeutikum zu. Daher gelten diese Grenzen auch für jede Charge des Referenzarzneimittels.
Von einer gleichen Wirksamkeit kann ausgegangen werden, wenn beim Biosimilar Moleküle und Stoffe mit derselben Affinität aneinander binden, mit derselben Potenz biologische Effekte ausgelöst werden und es dieselbe Pharmakokinetik wie das Referenzprodukt aufweist. Klinische Prüfungen müssen bei Biosimilars daher nur noch in der sensitivsten Population, d. h. in einer relevanten Indikation, durchgeführt werden. Dies muss der EMA als Zulassungsbehörde im Rahmen des Zulassungsverfahrens durch Daten nachgewiesen werden.
Was bedeutet eine „positive Opinion“?
Wird die Biosimilarität (der Wirkstoff ist dem des Referenzarzneimittels sehr ähnlich) durch umfassende Vergleichbarkeitsstudien und solide Daten zur pharmazeutischen Qualität nachgewiesen, bescheinigt die EMA dies mit einer „Positive Opinion“. Diese Beurteilung wird speziell auf jedes Biosimilar und den in ihm enthaltenen Wirkstoff zugeschnitten (Fall-zu-Fall-Bewertung).
Nach Erteilung der Zulassung für ein Biosimilar durch die Europäische Kommission, stellt ein besonderes Kontroll- und Überwachungssystem sicher, dass Sicherheit und Wirksamkeit des Produkts gewährleistet bleiben.
Seit die ersten Biosimilars 2006 in Europa zugelassen und auf den Markt gebracht wurden, ist kein einziger Fall bekannt, in dem ein Biosimilar aus Sicherheitsgründen oder mangelnder Wirksamkeit vom Markt genommen wurde [1]
[1].Kurki P, Barry S, Bourges I, Tsintili P, Wolff-Holz E. Safety, Immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: a regulatory perspective. Drugs. 2021;81:1881–1896. doi: 10.1007/s40265-021-01601-21. [DOI]
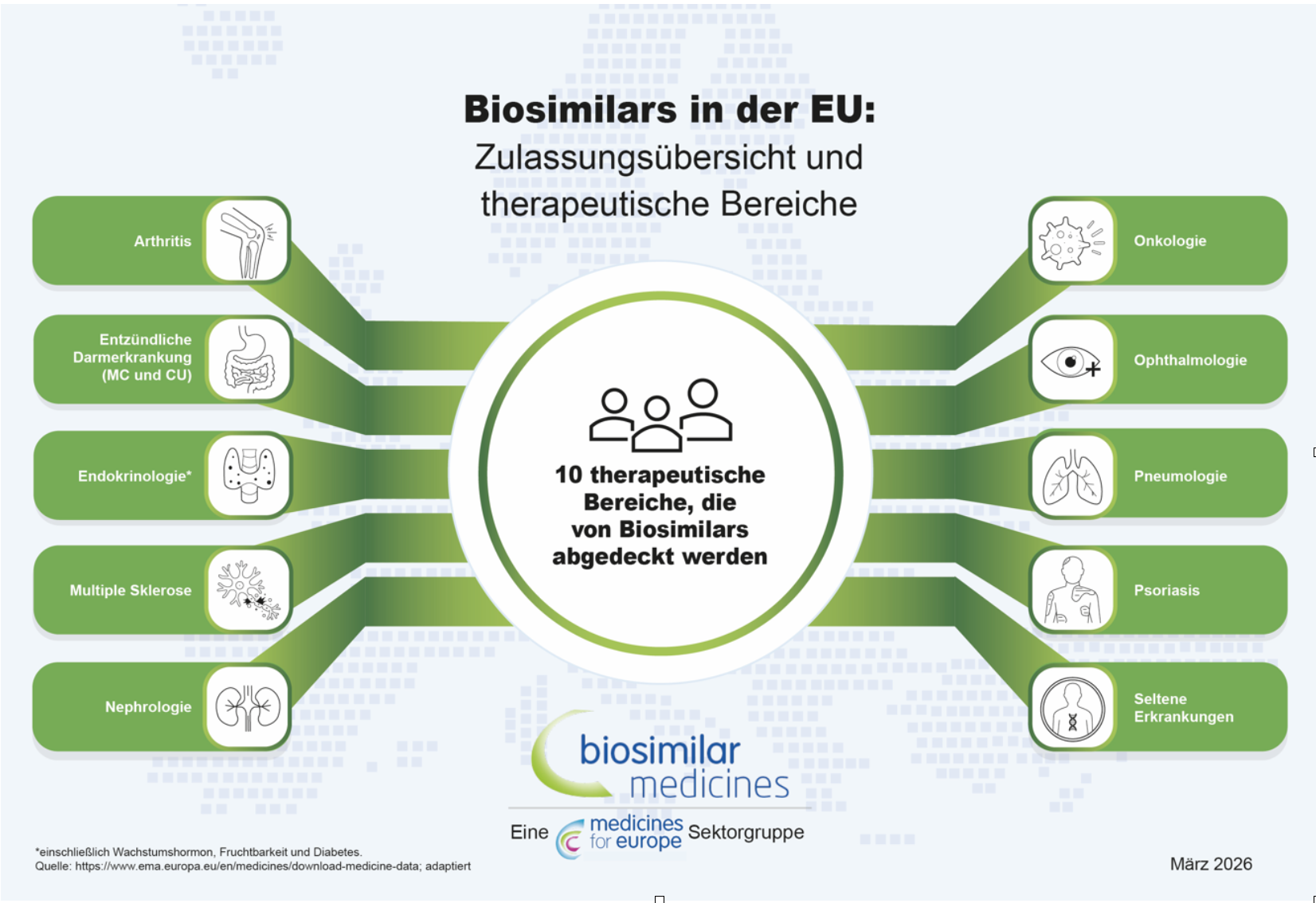
Zulassungen in Europa
Die Europäische Arzneimittel-Agentur (EMA) ist für die Prüfung und wissenschaftliche Bewertung von Biosimilars zuständig, während die Zulassung von Biosimilars durch die Europäische Kommission erfolgt. In Europa wurden bislang mehr als 160 Biosimilars in den therapeutischen Bereichen Arthritis, entzündliche Darmerkrankungen, Endokrinologie, Multiple Sklerose, Nephrologie, Onkologie, Ophthalmologie, Pneumologie, Psoriasis und seltene Erkrankungen zugelassen.